AMYOTROFICKÁ LATERÁLNÍ SKLERÓZA
Amyotrofická laterální skleróza (ALS) je neurodegenerativní onemocnění s roční incidencí 1-2/100 000, při němž dochází k postupnému úbytku motoneuronů v mozkové kůře, v jádrech hlavových nervů ve kmeni a v předních rozích míšních.
Klinické projevy zahrnují progresivní svalovou slabost, amyotrofii a spasticitu. První projevy nacházíme na končetinách nebo na bulbárním svalstvu, postupně dochází ke generalizaci onemocnění a rozvoji dechové. Poruchy čití a sfinkterů nepatří do obrazu ALS.
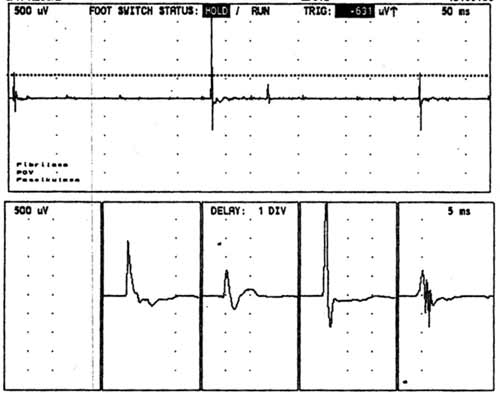
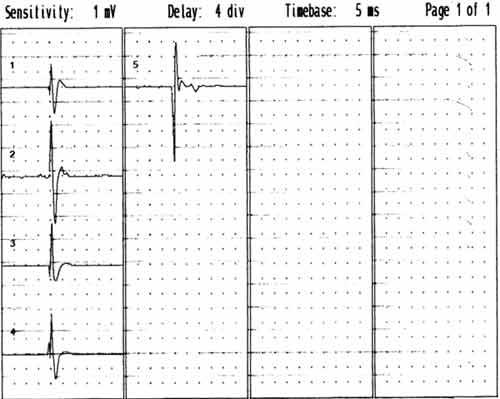
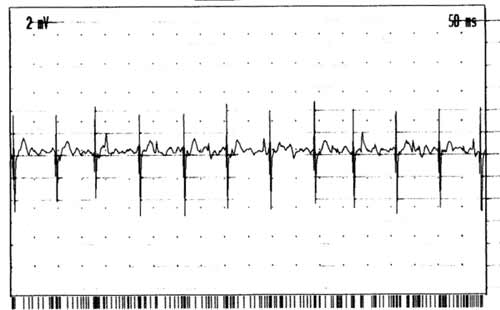
Diagnózu ALS potvrdí EMG
vyšetření. Dle standardů El Escorial je nutno prokázat léze centrálního a periferního motoneuronu
ve třech ze čtyř oblastí: mozkové nervy, cervikální, thorakální
a lumbální segmenty. Typický je nález četných, difúzně se vyskytujících
fascikulací na končetinách i na jazyku. V jehlové EMG ve svalech HK a DK
bývá chronický regenerační nález (pozitivní ostré vlny, fibrilace, vysoké
regenerační potenciály)
 |
 |
 |
ALS byla dlouho považována za onemocnění výlučně motorického systému. V posledních letech je ale stále větší pozornost věnována kognitivním změnám u těchto pacientů, v řadě případů se objevují poruchy frontálních exekutivních funkcí.
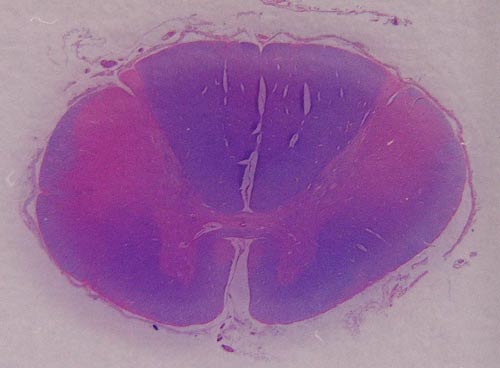
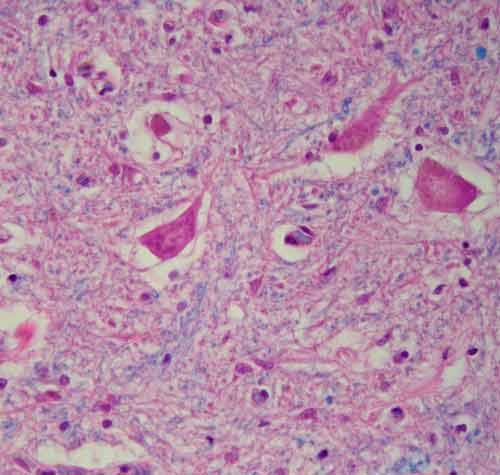
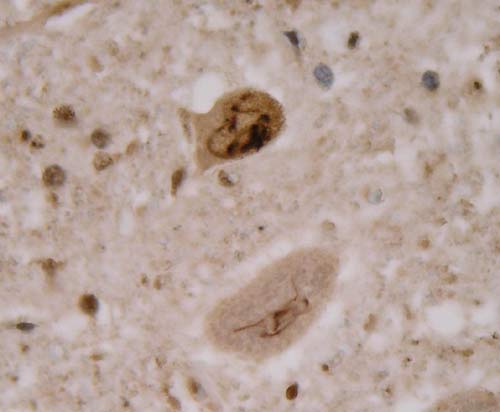
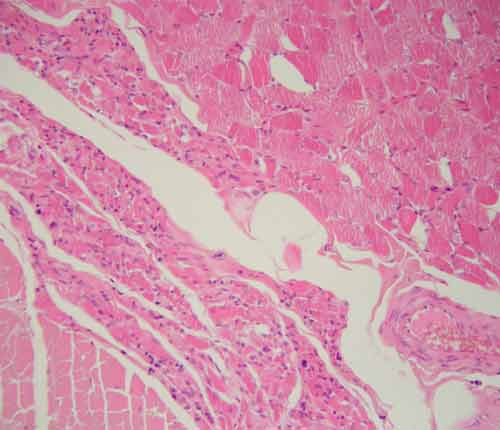
Hlavním neuropatologickým nálezem je numerická atrofie motorických neuronů v oblasti předních rohů míšních a v motorických jádrech hlavových nervů, skleróza a glióza postranních míšních provazců, atrofie kořenů míšních a neurogenní atrofie vláken příčně pruhované svaloviny. V regresivně změněných motorických neuronech bývají často přítomné různé typy diagnostických inkluzí.
 |
 |
 |
 |
Průběh onemocnění může zpomalit podávání glutamátového antagonisty riluzolu. Při poruchách polykání je metodou volby perkutánní endoskopická gastrostomie (PEG).
Asi 10% případů ALS je familiárních, u 40% postižených rodin byla nalezena mutace Cu-Zn superoxid-dismutázy (SOD1).
PRIMÁRNÍ LATERÁLNÍ SKLERÓZA
je diskutovaným onemocněním, o jehož existenci mnozí pochybují a považují
PLS za variantu ALS, jiní se domnívají, že se jedná o specifickou formu
postižení pouze centrálního motoneuronu (na rozdíl od ALS s poruchou prvního
i druhého motoneuronu).
Diagnóza PLS je čistě klinická, typický je začátek v dospělém věku, průběh je velmi pomalý, rozvoj spasticity s převahou postižení na DK trvá často řadu let a klinické postižení se omezuje na pyramidovou dráhu.
MRI může být normální nebo může zobrazit mírnou atrofii parietální krajiny, na motorických evokovaných potenciálech (MEP) mohu být prodloužené centrální časy vedení.
Progresivní bulbární paralýza
je další variantou ALS s postižením centrálního i periferního motoneuronu
jazyka a minimálním postižením končetinových svalů. Rovněž se může vyvinout
do obrazu klasické ALS.
Progresivní svalová atrofie
je varianta s asymetrickým pomalu se rozvíjejícím postižením periferního
motoneuronu oblasti cervikální, nebo lumbosakrální. V klinickém obraze
chybí postižení drah centrálního motoneuronu.